
Iron is essential for numerous biological processes, including oxygen transport, DNA synthesis, and cellular respiration. However, maintaining iron balance is crucial, as both iron deficiency and iron overload can lead to severe health issues. At the Laboratory of Iron Homeostasis, we investigate the mechanisms that regulate iron metabolism, focusing on iron recycling, sensing, and its impact on immune function and disease progression.
Research Summary
One aspect of our research focuses on iron recycling, mainly orchestrated by splenic red pulp macrophages (RPMs) that break down aging erythrocytes, releasing iron into the bloodstream. Despite being the primary iron source, limited knowledge exists about RPMs and the mechanisms influencing iron turnover efficacy. Our recent findings highlight the impairment of this process during aging. We demonstrated that age-related iron deposition in RPMs leads to their failure and demise, a challenge mitigated by dietary iron restriction in mice.
Another project explores unique functional and metabolic changes in RPMs in response to iron deficiency, aiding the organism's adjustment to restricted iron supplies. Concurrently, we investigate novel mechanisms in the sensing of body iron levels, particularly by specialized liver endothelial cells known as LSECs. We uncovered a new signaling pathway involving MAP kinases and the transcription factor ETS1, activated by excessive iron to amplify the expression of the well-known "iron sensor" Bmp6. Additionally, our research identifies LSECs as the primary cells removing free hemoglobin from circulation, playing a role in physiological iron recycling and hemoglobin detoxification under hemolytic conditions.
Scientific Impact
- - Identified impairment of iron recycling as an early hallmark of aging.
- - Discovered a new signaling pathway involved in iron sensing by liver endothelium.
- - Uncovered a previously unknown role of liver endothelium in the clearance of free hemoglobin, operating both under physiological and hemolytic conditions.
Future Goals
- - Explore the connections between iron recycling efficiency and splenic immune functions.
- - Deepen our understanding of the diverse roles of liver endothelium in maintaining iron homeostasis.
- - Further explore the adaptation of splenic macrophages to iron deficiency in health and disease.
Comment
"In our research, what we find particularly exciting is to realize that certain “dogmas” may only be partially accurate or even entirely false. This not only challenges existing beliefs but also creates opportunities to discover new principles in mammalian physiology that remain incompletely understood.", says Dr. Katarzyna Mleczko-Sanecka.
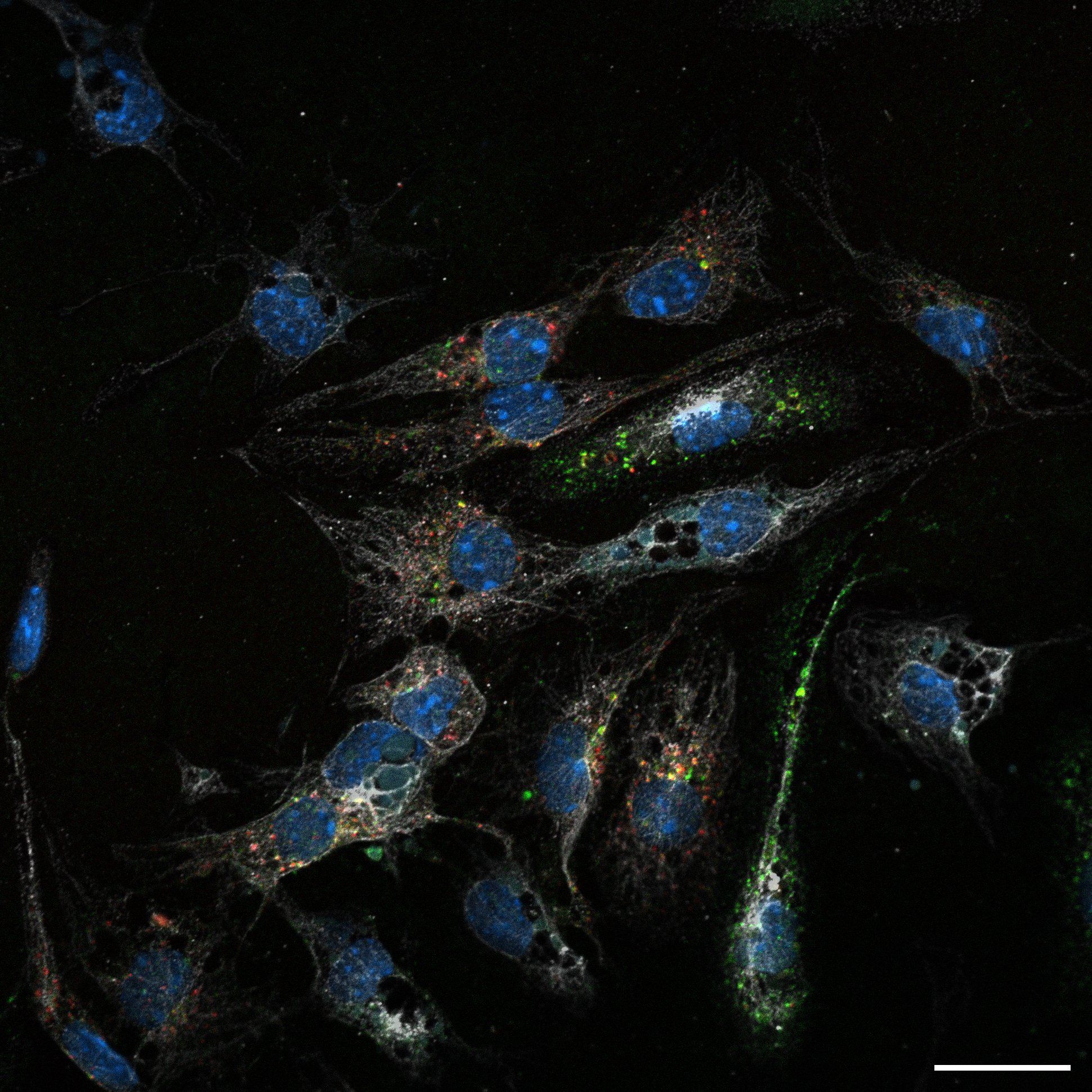
Primary murine LSECs performing uptake of fluorescentlylabeled hemoglobin. Illustration by Aneta Jończy.

