Rzadkie choroby neurologiczne, ataksje, objawy piramidowe, świadczące o uszkodzeniu ośrodków ruchowych i przejawiające się brakiem właściwej postawy ciała – to temat wywołujący duże zainteresowanie nie tylko wśród lekarzy i naukowców, lecz również pacjentów dotkniętych takimi schorzeniami i ich rodzin. Poszukiwanie przyczyn chorób rzadkich jest procesem długotrwałym i niełatwym, bowiem trudno jest uchwycić wszystkie molekularne aspekty, pozwalające na postawienie jednoznacznej odpowiedzi na pytanie: czy wykryta w badaniach genetycznych mutacja rzeczywiście wywołuje chorobę?
Ostatnie badania, przeprowadzone z wykorzystaniem modelu zwierzęcego przez zespół dra hab. Wojciecha Pokrzywy z Międzynarodowego Instytutu Biologii Molekularnej i Komórkowej w Warszawie (MIBMiK) wskazują, że mutacja konkretnego aminokwasu ważnego enzymu biorącego udział w degradacji białek może powodować zaburzenia neurorozwojowe.
Zespół badawczy, wykorzystując sekwencjonowanie eksomów do diagnozy pacjenta pediatrycznego z opóźnieniem rozwojowym, objawami piramidowymi i ataksją kończyn, zidentyfikował ultrarzadki wariant de novo zmiany sensu Asp126His w genie FEM1C kodującym ligazę ubikwityny – białko odpowiedzialne za rozpoznawanie innych białek skierowanych do degradacji przez komórkę i pośredniczące w ich usunięciu. – Wciąż nie było jednak jasne czy ta mutacja rzeczywiście odpowiada za chorobę pacjenta, co jest kluczowe przy stawianiu diagnozy i możliwości poszukiwań personalizowanych terapii – mówi dr hab. Wojciech Pokrzywa, Kierownik Laboratorium Metabolizmu Białek w MIBMiK.
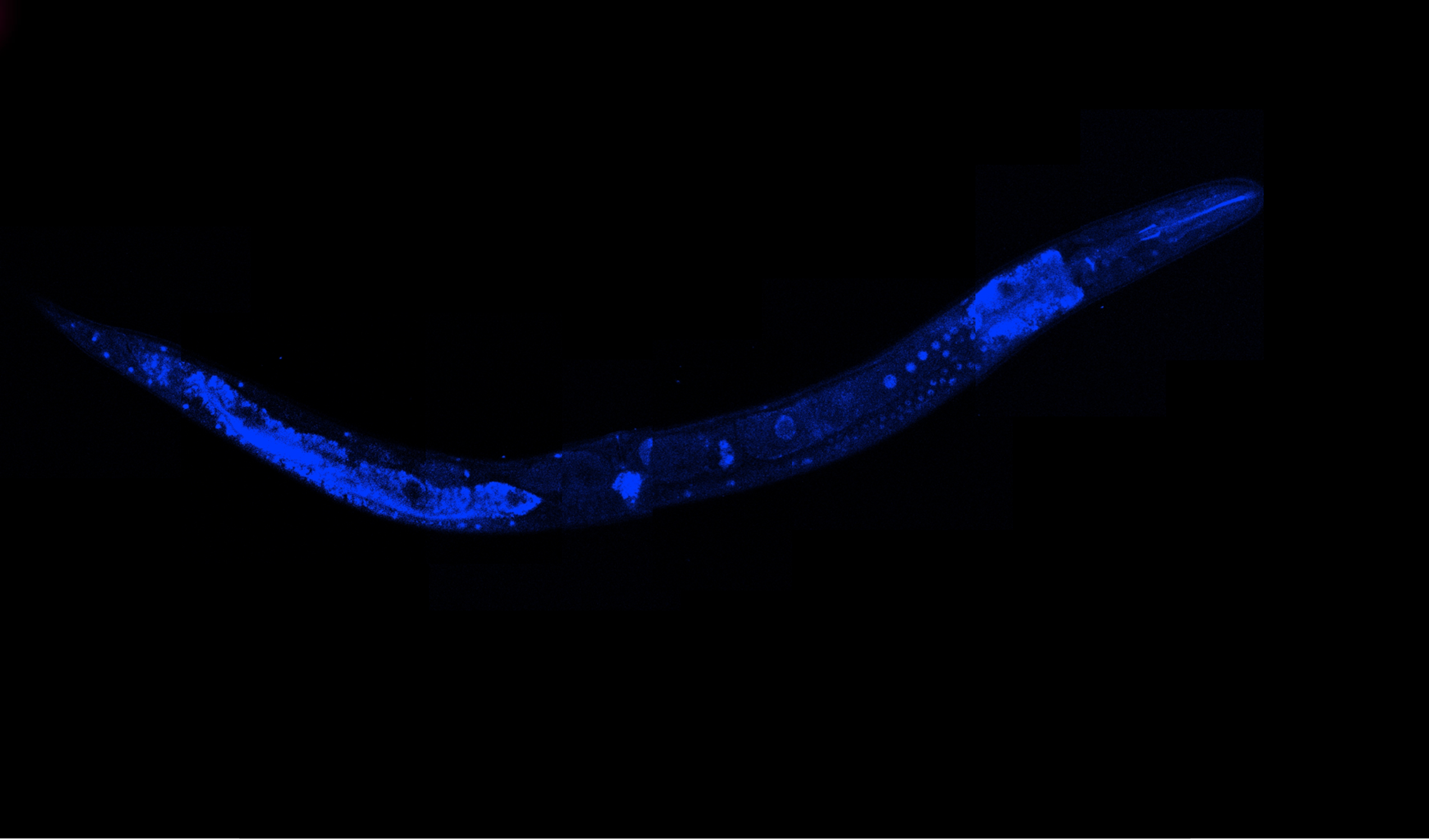
Wykonana przez zespół dra hab. Pokrzywy analiza bioinformatyczna wykazała, że wspomniana mutacja genu FEM1C zaburza wiązanie jego substratów białkowych przeznaczonych do degradacji, co w konsekwencji może prowadzić do ich akumulacji w komórce i toksycznego działania. Aby ocenić patogenność wariantu Asp126His w genie FEM1C, badacze wykorzystali nicienia Caenorhabditis elegans jako model choroby. W toku badań naukowcy stwierdzili, że zwierzęta wyrażające wariant analogiczny do mutacji pacjenta miały normalną architekturę mięśni, ale, podobnie jak pacjent, upośledzoną mobilność. Nicienie te były wrażliwe na inhibitor acetylocholinoesterazy, aldicarb, co wskazywało, że ich pogorszona lokomocja wynikała z nieprawidłowości synaptycznych, a nie dysfunkcji mięśni.
W wyniku przeprowadzonych badań przedstawione zostały pierwsze dowody z modelu zwierzęcego sugerujące, że mutacja w ewolucyjnie konserwowanym aminokwasie Asp126 w ligazie ubikwityny FEM1C powoduje zaburzenia neurorozwojowe u ludzi.
Wyniki badań opublikowane zostały w najnowszym wydaniu brytyjskiego czasopisma Human Molecular Genetics